Clonal analysis¶
[1]:
import cospar as cs
[2]:
cs.logging.print_version()
cs.settings.verbosity = 2 # range: 0 (error),1 (warning),2 (info),3 (hint).
cs.settings.set_figure_params(
format="png", figsize=[4, 3.5], dpi=75, fontsize=14, pointsize=3
)
Running cospar 0.3.0 (python 3.9.16) on 2023-07-14 11:53.
[3]:
# Each dataset should have its folder to avoid conflicts.
cs.settings.data_path = "data_cospar"
cs.settings.figure_path = "fig_cospar"
cs.hf.set_up_folders()
Load an existing dataset. (If you have pre-processed data, you can load it with cs.hf.read(file_name).)
[4]:
adata_orig = cs.datasets.hematopoiesis_subsampled()
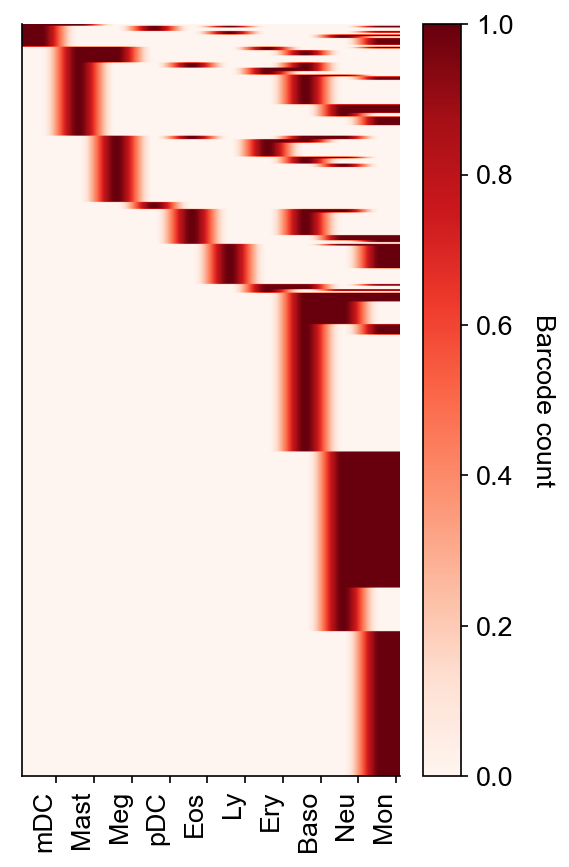
Show barcode heatmap as aggregated into given fate clusters (defined in adata_orig.obs['state_info'])
[5]:
selected_times = None
selected_fates = [
"Ccr7_DC",
"Mast",
"Meg",
"pDC",
"Eos",
"Lymphoid",
"Erythroid",
"Baso",
"Neutrophil",
"Monocyte",
]
celltype_names = ["mDC", "Mast", "Meg", "pDC", "Eos", "Ly", "Ery", "Baso", "Neu", "Mon"]
cs.pl.barcode_heatmap(
adata_orig,
selected_times=selected_times,
selected_fates=selected_fates,
color_bar=True,
rename_fates=celltype_names,
log_transform=False,
binarize=True,
)
Data saved at adata.uns['barcode_heatmap']
[5]:
<Axes: title={'center': '431 clones'}>
Fate coupling in the underlying clonal data, defined in our package as the normalized barcode covariance between cells annotated in different fates.
[6]:
cs.tl.fate_coupling(
adata_orig, selected_fates=selected_fates, source="X_clone"
) # compute the fate coupling
cs.pl.fate_coupling(adata_orig, source="X_clone") # actually plot the coupling
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
Results saved as dictionary at adata.uns['fate_coupling_X_clone']
[6]:
<Axes: title={'center': 'source: X_clone'}>

Fate hierarchy constructed from fate coupling of the underlying clonal data, using the neighbor-joining method.
[7]:
cs.tl.fate_hierarchy(
adata_orig, selected_fates=selected_fates, source="X_clone"
) # compute the fate hierarchy
cs.pl.fate_hierarchy(adata_orig, source="X_clone") # actually plot the hierarchy
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
normalize by X_clone
each cluster do not have a unique time point. Simply column-normalize the matrix
Results saved as dictionary at adata.uns['fate_hierarchy_X_clone']
/-Baso
/-|
/-| \-Mast
| |
/-| \-Eos
| |
| | /-Erythroid
/-| \-|
| | \-Meg
| |
| | /-Monocyte
--| \-|
| \-Neutrophil
|
| /-pDC
| /-|
\-| \-Ccr7_DC
|
\-Lymphoid
Next, we compute the clonal fate bias, -log(Q-value). We calculated a P-value that that a clone is enriched (or depleted) in a fate, using Fisher-Exact test (accounting for clone size). The P-value is then corrected to give a Q-value by Benjamini-Hochberg procedure. The alternative hypothesis options are: {‘two-sided’,‘greater’,‘less’}. The default is ‘two-sided’.
[8]:
cs.tl.clonal_fate_bias(
adata_orig, selected_fate="Monocyte", alternative="two-sided"
) # compute the fate hierarchy
cs.pl.clonal_fate_bias(adata_orig) # actually plot the hierarchy
100%|████████████████████████████████████████| 500/500 [00:00<00:00, 914.93it/s]
Data saved at adata.uns['clonal_fate_bias']
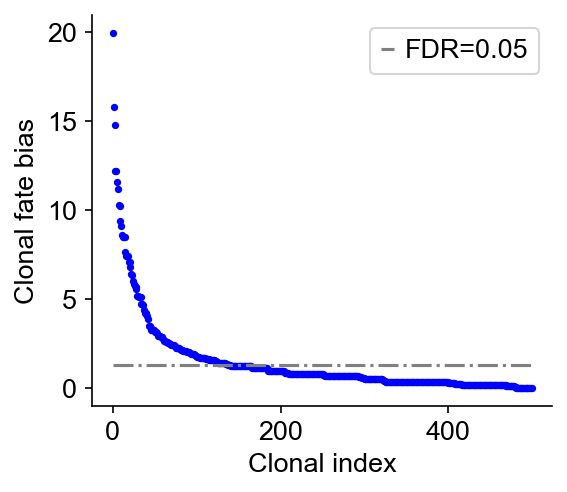
[9]:
result = adata_orig.uns["clonal_fate_bias"]
result
[9]:
| clone_id | clone_size | P_value | clonal_fraction_in_target_fate | Fate_bias | |
|---|---|---|---|---|---|
| 488 | 488 | 58 | 1.105140e-24 | 0.896552 | 23.956583 |
| 387 | 387 | 37 | 7.630927e-19 | 0.945946 | 18.117423 |
| 227 | 227 | 45 | 1.250056e-17 | 0.866667 | 16.903071 |
| 302 | 302 | 112 | 2.278106e-15 | 0.000000 | 14.642426 |
| 162 | 162 | 42 | 7.468800e-15 | 0.833333 | 14.126749 |
| ... | ... | ... | ... | ... | ... |
| 79 | 79 | 5 | 6.159379e-01 | 0.200000 | 0.210463 |
| 207 | 207 | 5 | 6.159379e-01 | 0.200000 | 0.210463 |
| 58 | 58 | 8 | 6.526613e-01 | 0.250000 | 0.185312 |
| 6 | 6 | 4 | 6.969634e-01 | 0.250000 | 0.156790 |
| 17 | 17 | 4 | 6.969634e-01 | 0.250000 | 0.156790 |
500 rows × 5 columns
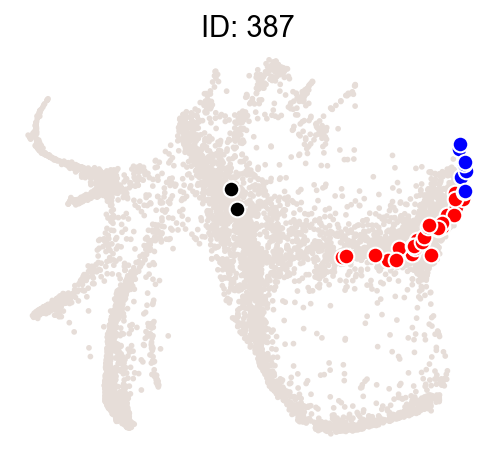
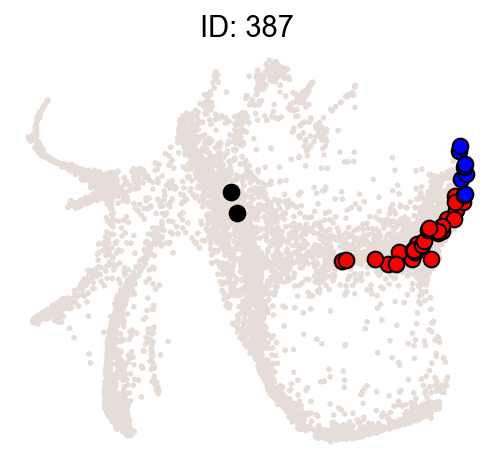
Illustrate some most biased clones.
[10]:
ids = result["clone_id"][:2]
cs.pl.clones_on_manifold(
adata_orig,
selected_clone_list=ids,
color_list=["black", "red", "blue"],
clone_markersize=15,
)
ERROR: XMLRPC request failed [code: -32500]
RuntimeError: PyPI no longer supports 'pip search' (or XML-RPC search). Please use https://pypi.org/search (via a browser) instead. See https://warehouse.pypa.io/api-reference/xml-rpc.html#deprecated-methods for more information.