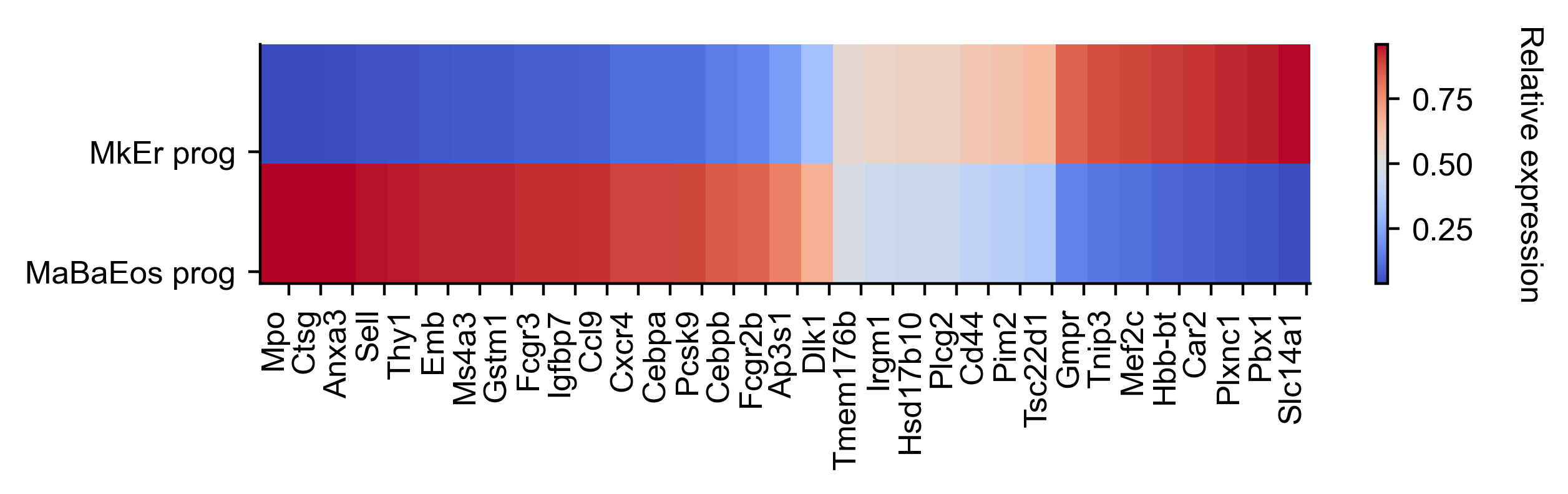Hematopoiesis¶
Hematopoiesis dataset from Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Science 367, (2020).
This dataset has 3 time points for both the clonal and state measurements. It has ~50000 clonally-labeled cells. Running the whole pipeline for the first time could take 5.2 hours in a standard personal computer. Most of the time (3.3 h) is used for generating the similarity matrix, which are saved for later usage.
Key components:
Part I: Infer transition map from all clonal data
Part II: Infer transition using end-point clones
Part III: Infer transition map from state information alone
Part IV: Predict fate bias for Gata1+ states
[1]:
import cospar as cs
import numpy as np
[2]:
cs.logging.print_version()
cs.settings.verbosity = 2
cs.settings.data_path = "LARRY_data" # A relative path to save data. If not existed before, create a new one.
cs.settings.figure_path = "LARRY_figure" # A relative path to save figures. If not existed before, create a new one.
cs.settings.set_figure_params(
format="png", figsize=[4, 3.5], dpi=75, fontsize=14, pointsize=2
)
Running cospar 0.2.0 (python 3.8.12) on 2022-02-07 21:21.
[3]:
# # This is for testing this notebook
# cs.settings.data_path='data_cospar'
# cs.settings.figure_path='fig_cospar'
# adata_orig=cs.datasets.hematopoiesis_subsampled()
# adata_orig.uns['data_des']=['LARRY_sp500_ranking1']
Loading data¶
[4]:
adata_orig = cs.datasets.hematopoiesis()
[5]:
adata_orig
[5]:
AnnData object with n_obs × n_vars = 49116 × 25289
obs: 'time_info', 'state_info'
uns: 'clonal_time_points', 'data_des', 'state_info_colors'
obsm: 'X_clone', 'X_emb', 'X_pca'
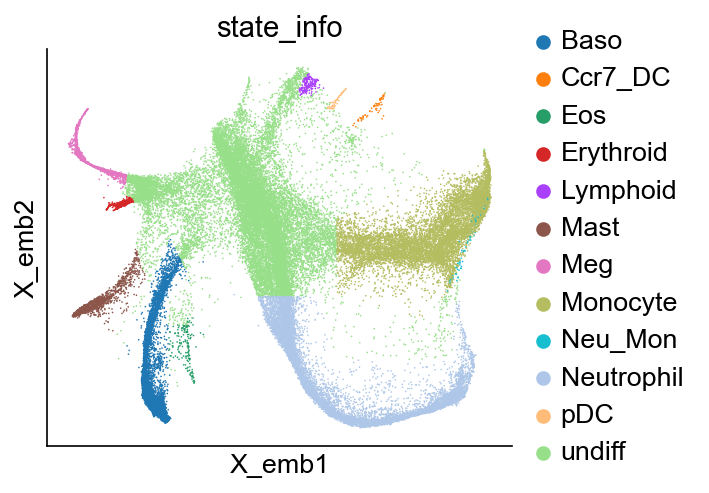
[6]:
cs.pl.embedding(adata_orig, color="state_info")
[7]:
cs.hf.check_available_choices(adata_orig)
Available transition maps: []
Available clusters: ['undiff', 'Meg', 'Mast', 'Neu_Mon', 'Eos', 'Erythroid', 'Ccr7_DC', 'Baso', 'Lymphoid', 'pDC', 'Neutrophil', 'Monocyte']
Available time points: ['2' '4' '6']
Clonal time points: ['2' '4' '6']
Basic clonal analysis¶
[8]:
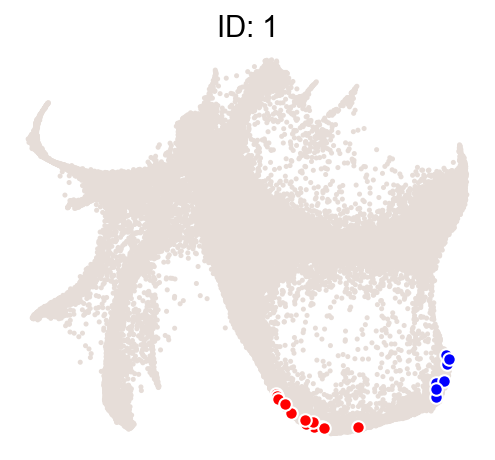
cs.pl.clones_on_manifold(
adata_orig, selected_clone_list=[1], color_list=["black", "red", "blue"]
)
[9]:
selected_times = "4"
selected_fates = [
"Ccr7_DC",
"Mast",
"Meg",
"pDC",
"Eos",
"Lymphoid",
"Erythroid",
"Baso",
"Neutrophil",
"Monocyte",
]
cs.tl.fate_coupling(
adata_orig,
source="X_clone",
selected_fates=selected_fates,
selected_times=selected_times,
normalize=False,
)
cs.pl.fate_coupling(adata_orig, source="X_clone")
Results saved as dictionary at adata.uns['fate_coupling_X_clone']
[9]:
<AxesSubplot:title={'center':'source: X_clone'}>

[10]:
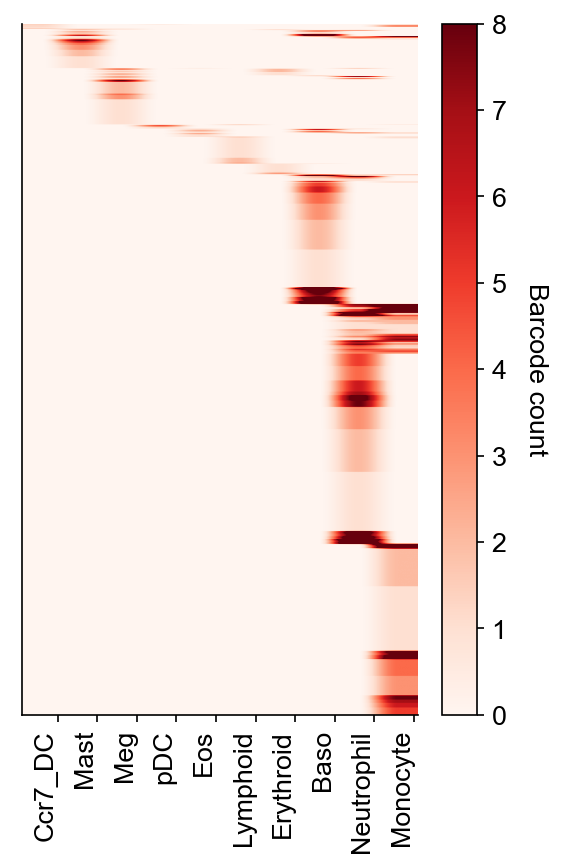
cs.pl.barcode_heatmap(
adata_orig,
selected_times=selected_times,
selected_fates=selected_fates,
color_bar=True,
)
Data saved at adata.uns['barcode_heatmap']
[10]:
<AxesSubplot:>
Part I: Infer transition map from all clonal data¶
Map inference¶
When first run, it takes around 3 h 37 mins, in which 3 h 20 mins are used for computing the similarity matrices and saving these data. It takes only 20 mins for later runs.
[11]:
adata = cs.tmap.infer_Tmap_from_multitime_clones(
adata_orig,
clonal_time_points=["2", "4", "6"],
later_time_point="6",
smooth_array=[20, 15, 10],
sparsity_threshold=0.2,
max_iter_N=3,
)
Trying to set attribute `.uns` of view, copying.
------Compute the full Similarity matrix if necessary------
------Infer transition map between initial time points and the later time one------
--------Current initial time point: 2--------
Step 1: Select time points
Number of multi-time clones post selection: 1216
Step 2: Optimize the transition map recursively
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.903
--------Current initial time point: 4--------
Step 1: Select time points
Number of multi-time clones post selection: 3047
Step 2: Optimize the transition map recursively
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.97
-----------Total used time: 1604.8337297439575 s ------------
Save pre-computed data (optional)¶
[12]:
save_data = False
if save_data:
cs.hf.save_map(adata)
Plotting¶
Transition profiles for single cells¶
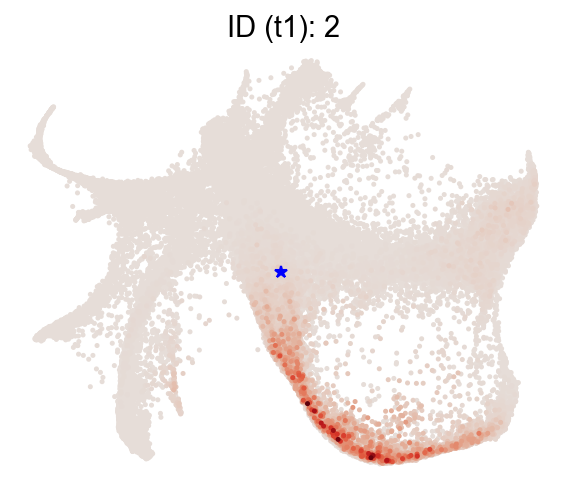
Forward transitions with map_backward=False.
[13]:
selected_state_id_list = [2]
cs.pl.single_cell_transition(
adata,
selected_state_id_list=selected_state_id_list,
color_bar=False,
source="transition_map",
map_backward=False,
)
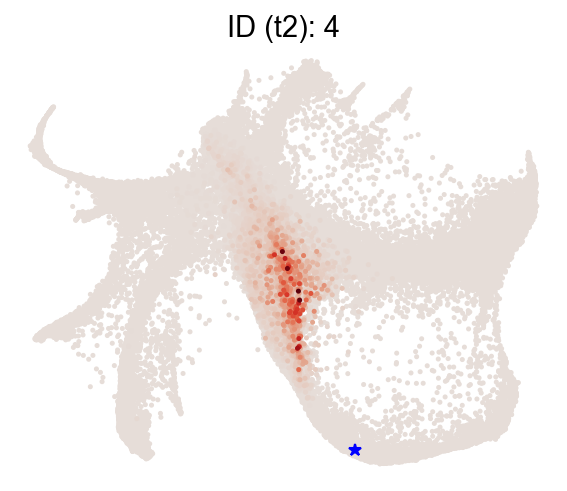
Backward transitions with map_backward=True.
[14]:
selected_state_id_list = [4]
cs.pl.single_cell_transition(
adata,
selected_state_id_list=selected_state_id_list,
color_bar=False,
source="transition_map",
map_backward=True,
)
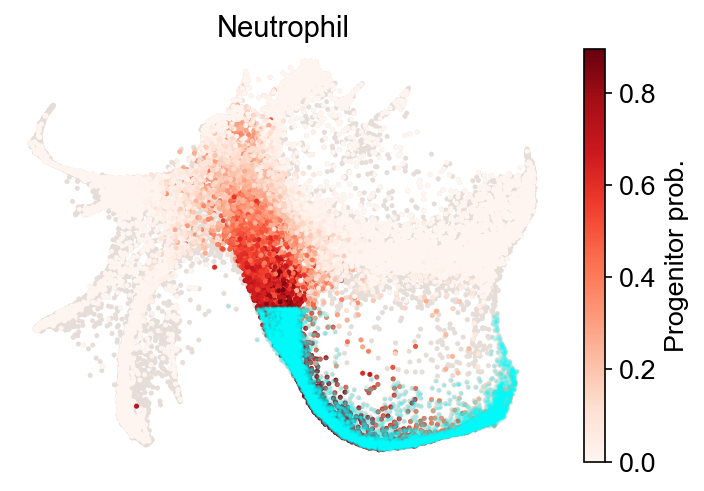
Fate map¶
[15]:
cs.tl.fate_map(
adata,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
map_backward=True,
)
cs.pl.fate_map(
adata,
selected_fates=["Neutrophil"],
source="transition_map",
plot_target_state=True,
show_histogram=False,
)
Results saved at adata.obs['fate_map_transition_map_Neutrophil']
Results saved at adata.obs['fate_map_transition_map_Monocyte']
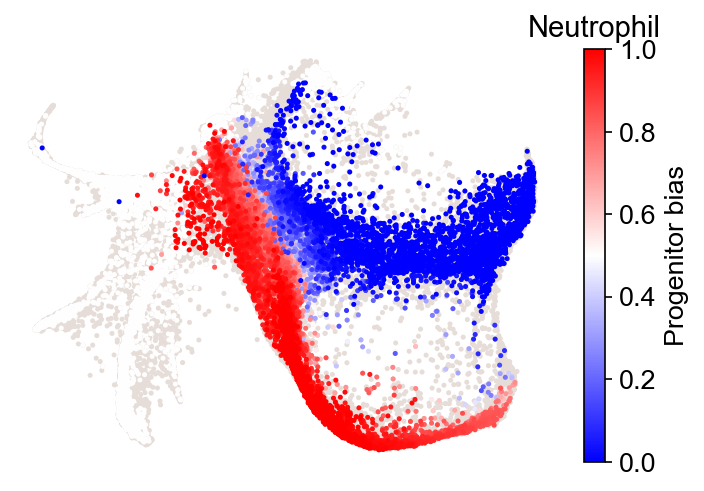
Fate bias¶
[16]:
cs.tl.fate_bias(
adata,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
pseudo_count=0,
sum_fate_prob_thresh=0.1,
)
cs.pl.fate_bias(
adata,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
plot_target_state=False,
selected_times=["4"],
)
Results saved at adata.obs['fate_map_transition_map_Neutrophil']
Results saved at adata.obs['fate_map_transition_map_Monocyte']
Results saved at adata.obs['fate_bias_transition_map_Neutrophil*Monocyte']
Fate coupling from the transition map¶
[17]:
selected_fates = [
"Ccr7_DC",
"Mast",
"Meg",
"pDC",
"Eos",
"Lymphoid",
"Erythroid",
"Baso",
"Neutrophil",
"Monocyte",
]
cs.tl.fate_coupling(adata, selected_fates=selected_fates, source="transition_map")
cs.pl.fate_coupling(adata, source="transition_map")
Results saved as dictionary at adata.uns['fate_coupling_transition_map']
[17]:
<AxesSubplot:title={'center':'source: transition_map'}>

Hierarchy¶
[18]:
cs.tl.fate_hierarchy(adata, selected_fates=selected_fates, source="transition_map")
cs.pl.fate_hierarchy(adata, source="transition_map")
Results saved as dictionary at adata.uns['fate_hierarchy_transition_map']
/-Erythroid
/-|
/-| \-Meg
| |
/-| \-Mast
| |
| | /-Baso
| \-|
--| \-Eos
|
| /-Monocyte
| /-|
| | \-Neutrophil
\-|
| /-Lymphoid
| /-|
\-| \-Ccr7_DC
|
\-pDC
Dynamic trajectory inference¶
[19]:
cs.tl.progenitor(
adata,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
map_backward=True,
bias_threshold_A=0.5,
bias_threshold_B=0.5,
sum_fate_prob_thresh=0.2,
avoid_target_states=True,
)
cs.pl.progenitor(
adata, selected_fates=["Neutrophil", "Monocyte"], source="transition_map"
)
Results saved at adata.obs['fate_map_transition_map_Neutrophil']
Results saved at adata.obs['fate_map_transition_map_Monocyte']
Results saved at adata.obs['fate_bias_transition_map_Neutrophil*Monocyte']
Results saved at adata.obs[f'progenitor_transition_map_Neutrophil'] and adata.obs[f'diff_trajectory_transition_map_Neutrophil']
Results saved at adata.obs[f'progenitor_transition_map_Monocyte'] and adata.obs[f'diff_trajectory_transition_map_Monocyte']
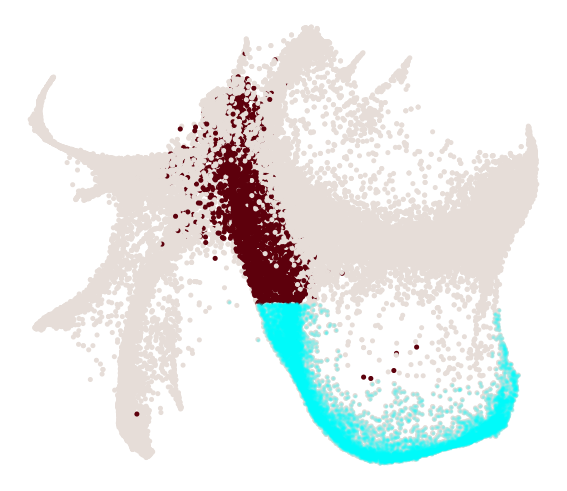

Gene trend along the dynamic trajectory¶
[20]:
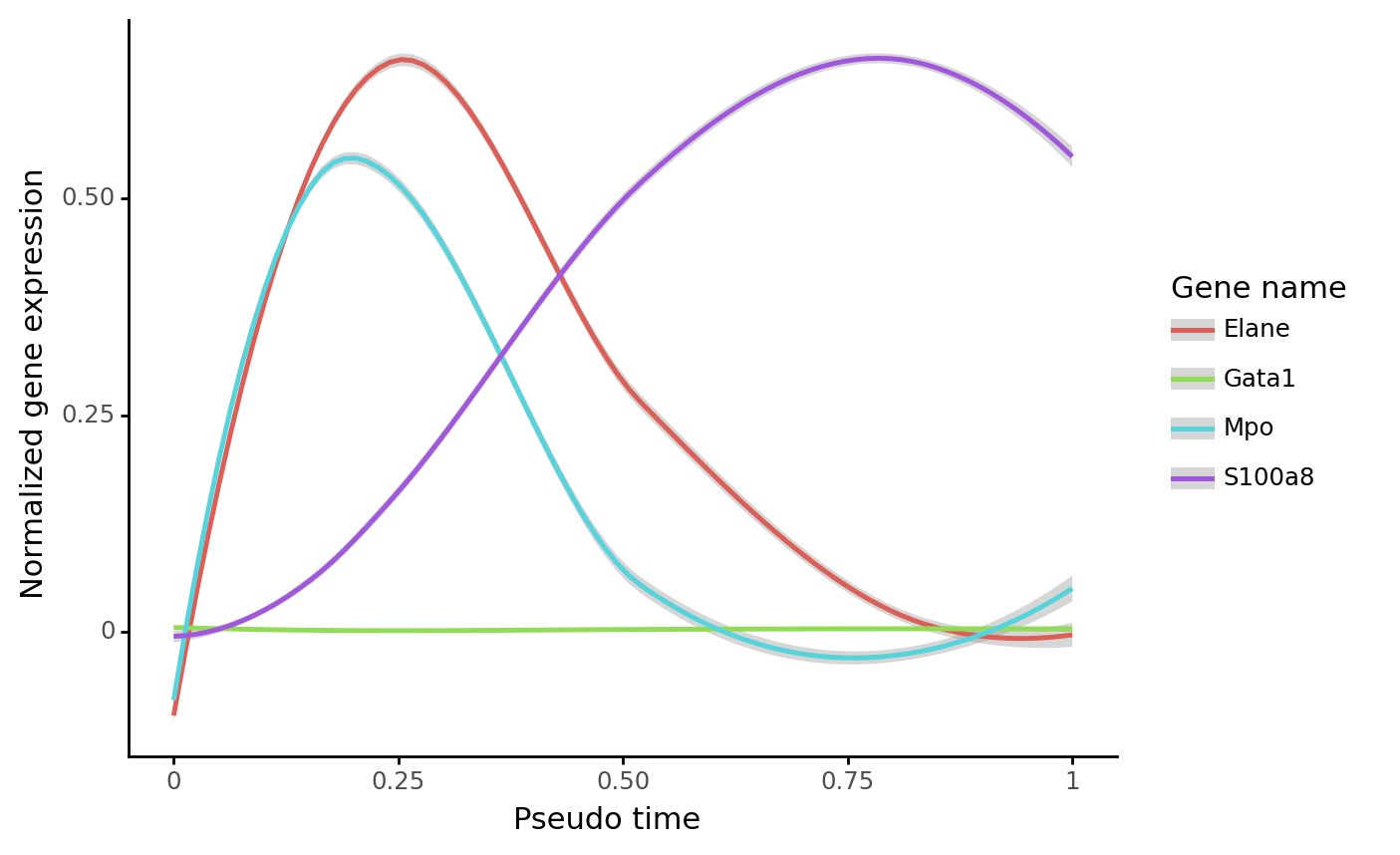
gene_name_list = ["Gata1", "Mpo", "Elane", "S100a8"]
selected_fate = "Neutrophil"
cs.pl.gene_expression_dynamics(
adata, selected_fate, gene_name_list, traj_threshold=0.2, invert_PseudoTime=False
)
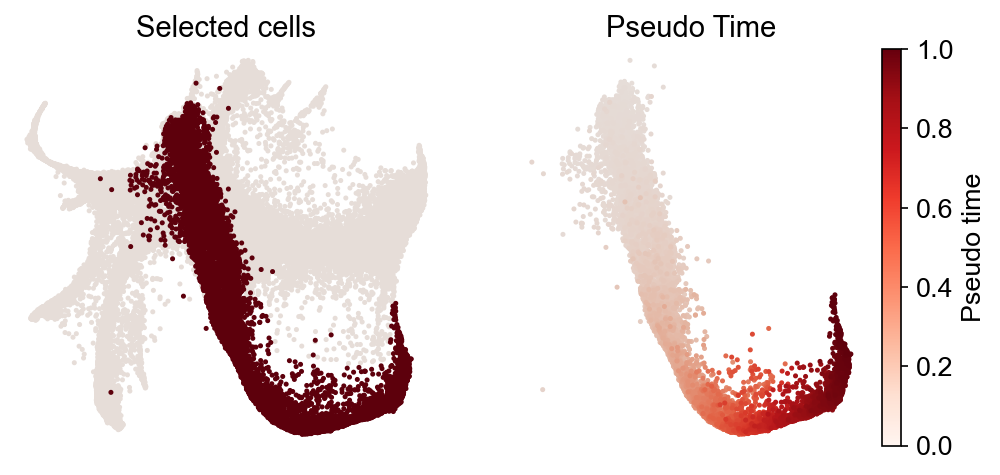
Part II: Infer transition map from end-point clones¶
When run for the firs time, assuming that the similarity matrices are pre-computed, it takes 73 mins. Around 40 mins of which are used to compute the initialized map.
We group time points ‘2’ and ‘4’ so that the inference on day 2 states (which has much fewer cells) are better.
[21]:
time_info = np.array(adata_orig.obs["time_info"])
time_info[time_info == "2"] = "24"
time_info[time_info == "4"] = "24"
adata_orig.obs["time_info"] = time_info
adata_orig = cs.pp.initialize_adata_object(adata_orig)
Clones without any cells are removed.
Time points with clonal info: ['24' '6']
WARNING: Default ascending order of time points are: ['24' '6']. If not correct, run cs.hf.update_time_ordering for correction.
WARNING: Please make sure that the count matrix adata.X is NOT log-transformed.
[22]:
adata_1 = cs.tmap.infer_Tmap_from_one_time_clones(
adata_orig,
initial_time_points=["24"],
later_time_point="6",
initialize_method="OT",
OT_cost="GED",
smooth_array=[20, 15, 10],
max_iter_N=[1, 3],
sparsity_threshold=0.2,
use_full_Smatrix=True,
)
Trying to set attribute `.uns` of view, copying.
Trying to set attribute `.uns` of view, copying.
--------Infer transition map between initial time points and the later time one-------
--------Current initial time point: 24--------
Step 0: Pre-processing and sub-sampling cells-------
Step 1: Use OT method for initialization-------
Load pre-computed custom OT matrix
Step 2: Jointly optimize the transition map and the initial clonal states-------
-----JointOpt Iteration 1: Infer initial clonal structure
-----JointOpt Iteration 1: Update the transition map by CoSpar
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.974
Convergence (JointOpt, iter_N=1): corr(previous_T, current_T)=0.144
Finishing Joint Optimization, used time 1308.7951350212097
-----------Total used time: 1341.8913249969482 s ------------
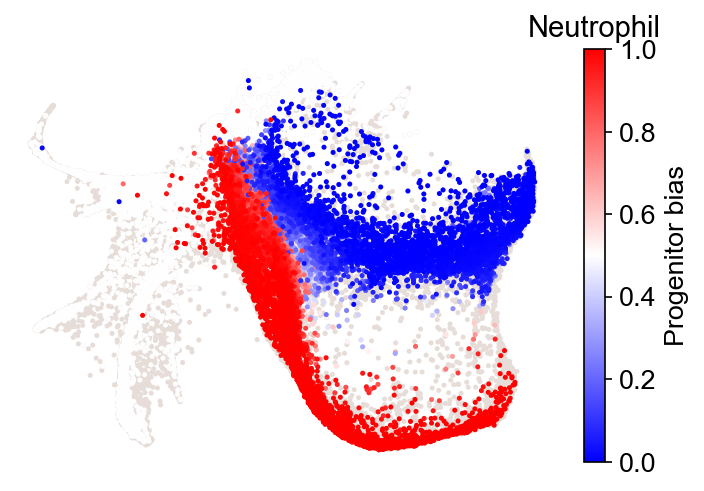
Fate bias¶
[23]:
cs.tl.fate_bias(
adata_1,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
pseudo_count=0,
sum_fate_prob_thresh=0.1,
)
cs.pl.fate_bias(
adata_1,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
plot_target_state=False,
)
Results saved at adata.obs['fate_map_transition_map_Neutrophil']
Results saved at adata.obs['fate_map_transition_map_Monocyte']
Results saved at adata.obs['fate_bias_transition_map_Neutrophil*Monocyte']
Part III: Infer transition map from state information alone¶
[24]:
adata_2 = cs.tmap.infer_Tmap_from_state_info_alone(
adata_orig,
initial_time_points=["24"],
later_time_point="6",
initialize_method="OT",
OT_cost="GED",
smooth_array=[20, 15, 10],
max_iter_N=[1, 3],
sparsity_threshold=0.2,
use_full_Smatrix=True,
)
Step I: Generate pseudo clones where each cell has a unique barcode-----
Trying to set attribute `.uns` of view, copying.
Step II: Perform joint optimization-----
Trying to set attribute `.uns` of view, copying.
--------Infer transition map between initial time points and the later time one-------
--------Current initial time point: 24--------
Step 0: Pre-processing and sub-sampling cells-------
Step 1: Use OT method for initialization-------
Load pre-computed custom OT matrix
Step 2: Jointly optimize the transition map and the initial clonal states-------
-----JointOpt Iteration 1: Infer initial clonal structure
-----JointOpt Iteration 1: Update the transition map by CoSpar
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.977
Convergence (JointOpt, iter_N=1): corr(previous_T, current_T)=0.14
Finishing Joint Optimization, used time 1854.5912249088287
-----------Total used time: 1923.1848938465118 s ------------
[25]:
cs.tl.fate_bias(
adata_2,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
pseudo_count=0,
sum_fate_prob_thresh=0.1,
)
cs.pl.fate_bias(
adata_2,
selected_fates=["Neutrophil", "Monocyte"],
source="transition_map",
plot_target_state=False,
)
Results saved at adata.obs['fate_map_transition_map_Neutrophil']
Results saved at adata.obs['fate_map_transition_map_Monocyte']
Results saved at adata.obs['fate_bias_transition_map_Neutrophil*Monocyte']

Part IV: Predict fate bias for Gata1+ states¶
Load data¶
We load a dataset that also includes non-clonally labeled cells in the Gata1+ states (in total ~38K cells), which increases the statistical power of DGE analysis.
[26]:
cs.settings.data_path = "LARRY_data_Gata1" # A relative path to save data. If not existed before, create a new one.
cs.settings.figure_path = "LARRY_figure_Gata1" # A relative path to save figures. If not existed before, create a new one.
adata_orig_1 = cs.datasets.hematopoiesis_Gata1_states()
[27]:
adata_orig_1 = cs.pp.initialize_adata_object(adata_orig_1)
Time points with clonal info: ['2' '4' '6']
WARNING: Default ascending order of time points are: ['2' '4' '6']. If not correct, run cs.hf.update_time_ordering for correction.
WARNING: Please make sure that the count matrix adata.X is NOT log-transformed.
[28]:
cs.pl.embedding(adata_orig_1, color="time_info")
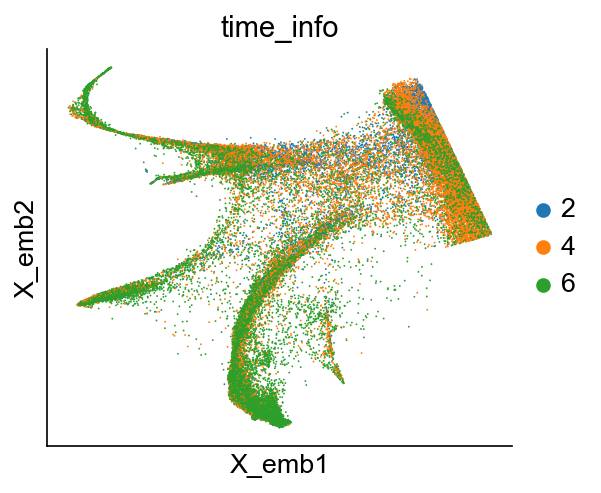
[29]:
adata_orig_1
[29]:
AnnData object with n_obs × n_vars = 38457 × 25289
obs: 'n_counts', 'Source', 'Well', 'mito_frac', 'time_info', 'state_info'
var: 'highly_variable'
uns: 'AllCellCyc_genes', 'clonal_time_points', 'data_des', 'max_mito', 'min_tot', 'new_highvar_genes', 'state_info_colors', 'time_info_colors', 'time_ordering'
obsm: 'X_clone', 'X_emb', 'X_pca', 'X_umap'
[30]:
## This is for testing this notebook
# adata_orig_1=adata_orig
# adata_3=adata_1
Infer transition map from all clones¶
[31]:
adata_3 = cs.tmap.infer_Tmap_from_multitime_clones(
adata_orig_1,
later_time_point="6",
smooth_array=[20, 15, 10],
sparsity_threshold=0.1,
intraclone_threshold=0.1,
extend_Tmap_space=True,
)
------Compute the full Similarity matrix if necessary------
Trying to set attribute `.uns` of view, copying.
------Infer transition map between initial time points and the later time one------
--------Current initial time point: 2--------
Step 1: Select time points
Number of multi-time clones post selection: 355
Step 2: Optimize the transition map recursively
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.969
--------Current initial time point: 4--------
Step 1: Select time points
Number of multi-time clones post selection: 1270
Step 2: Optimize the transition map recursively
Load pre-computed similarity matrix
Iteration 1, Use smooth_round=20
Iteration 2, Use smooth_round=15
Iteration 3, Use smooth_round=10
Convergence (CoSpar, iter_N=3): corr(previous_T, current_T)=0.985
-----------Total used time: 477.8539500236511 s ------------
Fate bias¶
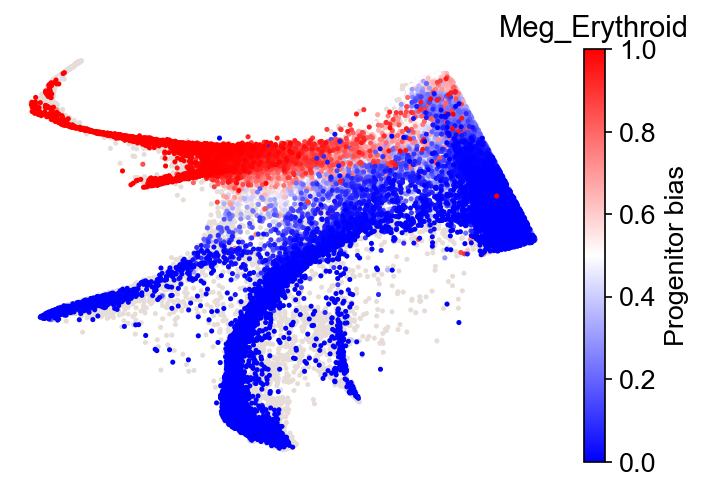
[32]:
selected_fates = [["Meg", "Erythroid"], ["Baso", "Mast", "Eos"]]
cs.tl.fate_bias(
adata_3,
selected_fates=selected_fates,
source="transition_map",
pseudo_count=0,
sum_fate_prob_thresh=0.05,
)
cs.pl.fate_bias(
adata_3,
selected_fates=selected_fates,
source="transition_map",
plot_target_state=False,
)
Results saved at adata.obs['fate_map_transition_map_Meg_Erythroid']
Results saved at adata.obs['fate_map_transition_map_Baso_Mast_Eos']
Results saved at adata.obs['fate_bias_transition_map_Meg_Erythroid*Baso_Mast_Eos']
Identify ancestor populations¶
[33]:
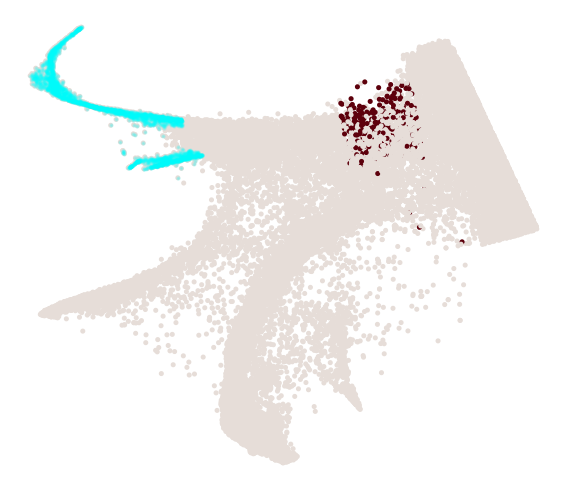
x_emb = adata_orig_1.obsm["X_emb"][:, 0]
y_emb = adata_orig_1.obsm["X_emb"][:, 1]
index_2 = cs.hf.above_the_line(adata_orig_1.obsm["X_emb"], [0, -500], [-300, 1000])
index_2_2 = cs.hf.above_the_line(adata_orig_1.obsm["X_emb"], [-450, -600], [-600, 800])
final_mask = (index_2_2) & (~index_2) # & index_3 & index_4 & index_5 #mask_1 &
cs.pl.customized_embedding(x_emb, y_emb, final_mask)
[33]:
<AxesSubplot:>

[34]:
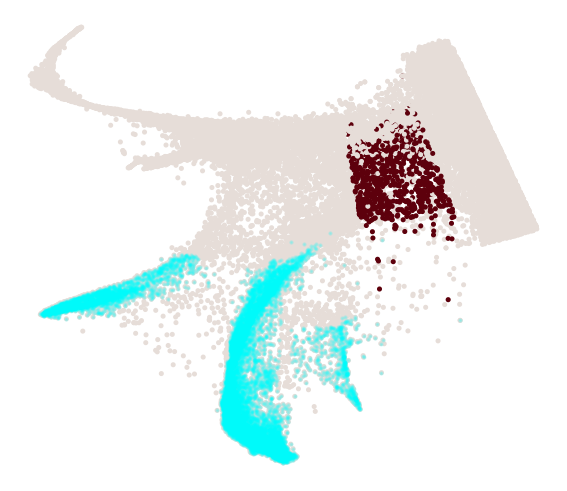
selected_fates = [["Meg", "Erythroid"], ["Baso", "Mast", "Eos"]]
cs.tl.progenitor(
adata_3,
selected_fates=selected_fates,
source="transition_map",
map_backward=True,
bias_threshold_A=0.6,
bias_threshold_B=0.3,
sum_fate_prob_thresh=0.05,
avoid_target_states=True,
)
cs.pl.progenitor(
adata_3, selected_fates=selected_fates, source="transition_map", mask=final_mask
)
Results saved at adata.obs['fate_map_transition_map_Meg_Erythroid']
Results saved at adata.obs['fate_map_transition_map_Baso_Mast_Eos']
Results saved at adata.obs['fate_bias_transition_map_Meg_Erythroid*Baso_Mast_Eos']
Results saved at adata.obs[f'progenitor_transition_map_Meg_Erythroid'] and adata.obs[f'diff_trajectory_transition_map_Meg_Erythroid']
Results saved at adata.obs[f'progenitor_transition_map_Baso_Mast_Eos'] and adata.obs[f'diff_trajectory_transition_map_Baso_Mast_Eos']
DGE analysis¶
[35]:
cell_group_A = np.array(adata_3.obs[f"progenitor_transition_map_Meg_Erythroid"])
cell_group_B = np.array(adata_3.obs[f"progenitor_transition_map_Baso_Mast_Eos"])
dge_gene_A, dge_gene_B = cs.tl.differential_genes(
adata_3, cell_group_A=cell_group_A, cell_group_B=cell_group_B, FDR_cutoff=0.05
)
[36]:
# All, ranked, DGE genes for group A
dge_gene_A
[36]:
| index | gene | Qvalue | mean_1 | mean_2 | ratio | |
|---|---|---|---|---|---|---|
| 0 | 4 | Ctsg | 0.000000e+00 | 0.103370 | 2.567811 | -1.693123 |
| 1 | 3 | Emb | 0.000000e+00 | 0.186135 | 2.703714 | -1.642705 |
| 2 | 26 | Elane | 3.264509e-241 | 0.151518 | 2.530409 | -1.616299 |
| 3 | 2 | Plac8 | 0.000000e+00 | 1.096986 | 4.811569 | -1.470611 |
| 4 | 17 | Igfbp4 | 0.000000e+00 | 1.042382 | 3.969988 | -1.282990 |
| ... | ... | ... | ... | ... | ... | ... |
| 965 | 2772 | Rps29 | 2.823620e-02 | 2.905993 | 3.008932 | -0.037529 |
| 966 | 2343 | Rpl38 | 8.297305e-03 | 5.915980 | 6.061140 | -0.029967 |
| 967 | 2976 | Rpl5 | 4.697313e-02 | 4.714540 | 4.806636 | -0.023065 |
| 968 | 2544 | Ly6e | 1.556172e-02 | 2.882883 | 2.944137 | -0.022582 |
| 969 | 2705 | Isg20 | 2.485104e-02 | 0.330973 | 0.342714 | -0.012671 |
970 rows × 6 columns
[37]:
sub_predicted_upper = [
"Slc14a1",
"Plxnc1",
"Mef2c",
"Tnip3",
"Pbx1",
"Hbb-bt",
"Tmem176b",
"Car2",
"Pim2",
"Tsc22d1",
"Plcg2",
"Gmpr",
"Cd44",
"Irgm1",
"Hsd17b10",
"Dlk1",
]
sub_predicted_lower = [
"Ctsg",
"Emb",
"Ccl9",
"Igfbp7",
"Ap3s1",
"Gstm1",
"Ms4a3",
"Fcgr3",
"Mpo",
"Cebpb",
"Thy1",
"Anxa3",
"Sell",
"Pcsk9",
"Fcgr2b",
"Cebpa",
"Cxcr4",
]
[38]:
state_info = np.array(adata_3.obs["state_info"])
state_info[cell_group_A > 0] = "MegEr_Prog"
state_info[cell_group_B > 0] = "EMB_Prog"
[39]:
adata_3.obs["state_info"] = state_info
cs.pl.embedding(adata_3, color="state_info")
/Users/shouwenwang/miniconda3/envs/CoSpar_test/lib/python3.8/site-packages/anndata/_core/anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Reordering categories will always return a new Categorical object.
... storing 'state_info' as categorical

[40]:
cs.settings.set_figure_params(
format="pdf", figsize=[4, 3.5], dpi=200, fontsize=10, pointsize=3
)
gene_list = sub_predicted_upper + sub_predicted_lower
selected_fates = ["MegEr_Prog", "EMB_Prog"]
renames = ["MkEr prog", "MaBaEos prog"]
gene_expression_matrix = cs.pl.gene_expression_heatmap(
adata_3,
selected_genes=gene_list,
selected_fates=selected_fates,
rename_fates=renames,
horizontal=True,
fig_width=6.5,
fig_height=2,
)