Preprocessing¶
Note that this step is optional. If the data do not yet have one of X_pca, X_emb, or state_info, you will need to run the preprocessing and dimension reduction.
[2]:
import os
[3]:
import cospar as cs
[4]:
cs.logging.print_version()
cs.settings.verbosity = 2 # range: 0 (error),1 (warning),2 (info),3 (hint).
cs.settings.set_figure_params(
format="png", figsize=[4, 3.5], dpi=75, fontsize=14, pointsize=3
)
Running cospar 0.3.0 (python 3.9.16) on 2023-07-14 11:59.
ERROR: XMLRPC request failed [code: -32500]
RuntimeError: PyPI no longer supports 'pip search' (or XML-RPC search). Please use https://pypi.org/search (via a browser) instead. See https://warehouse.pypa.io/api-reference/xml-rpc.html#deprecated-methods for more information.
[5]:
# Each dataset should have its folder to avoid conflicts.
cs.settings.data_path = "data_cospar"
cs.settings.figure_path = "fig_cospar"
cs.hf.set_up_folders()
Load an existing dataset. (If you have pre-processed data, you can load it with cs.hf.read(file_name).)
[6]:
adata_orig = cs.datasets.hematopoiesis_subsampled()
Dimension reduction¶
Select highly variable genes. Before this, there is count normalization. It requires that the count matrix is NOT log-normalized.
[7]:
cs.pp.get_highly_variable_genes(
adata_orig,
normalized_counts_per_cell=10000,
min_counts=3,
min_cells=3,
min_gene_vscore_pctl=90,
)
Finding highly variable genes...
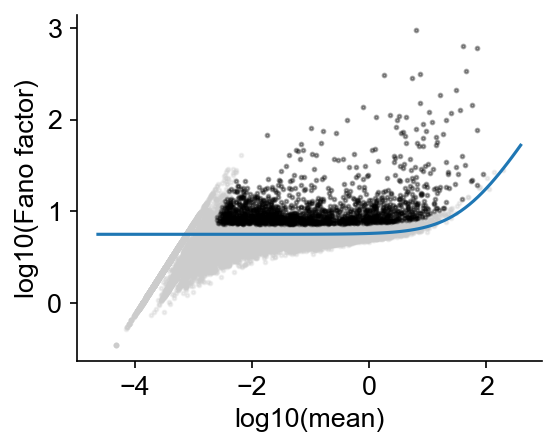
Keeping 1615 genes
Compute for each gene its correlation with a set of cell cycle genes. The default cell cycle genes are for mouse. You need to use your own genes for a different species. This step is optional, but recommended.
[8]:
cs.pp.remove_cell_cycle_correlated_genes(
adata_orig,
cycling_gene_list=[
"Ube2c",
"Hmgb2",
"Hmgn2",
"Tuba1b",
"Ccnb1",
"Tubb5",
"Top2a",
"Tubb4b",
],
)
adata.var['highly_variable'] not updated.
Please choose corr_threshold properly, and set confirm_change=True
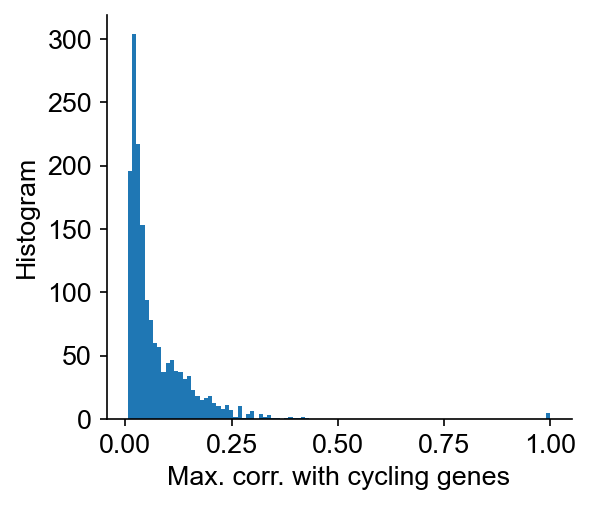
Now, confirm the change at a specific cutoff threshold.
[9]:
cs.pp.remove_cell_cycle_correlated_genes(
adata_orig, corr_threshold=0.2, confirm_change=True
)
Number of selected non-cycling highly variable genes: 1508
Remove 107 cell cycle correlated genes.
adata.var['highly_variable'] updated
Compute the X_pca, X_emb. X_pca will be used to build the similarity matrix later. X_emb is only used for visualization. You can also pass your favorite embedding directly to adata.obsm['X_emb'].
[10]:
cs.pp.get_X_pca(adata_orig, n_pca_comp=40)
cs.pp.get_X_emb(adata_orig, n_neighbors=20, umap_min_dist=0.3)
WARNING: get_X_pca assumes that the count matrix adata.X is NOT log-transformed.
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/validation.py:727: FutureWarning: np.matrix usage is deprecated in 1.0 and will raise a TypeError in 1.2. Please convert to a numpy array with np.asarray. For more information see: https://numpy.org/doc/stable/reference/generated/numpy.matrix.html
warnings.warn(
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/validation.py:727: FutureWarning: np.matrix usage is deprecated in 1.0 and will raise a TypeError in 1.2. Please convert to a numpy array with np.asarray. For more information see: https://numpy.org/doc/stable/reference/generated/numpy.matrix.html
warnings.warn(
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/extmath.py:152: RuntimeWarning: invalid value encountered in matmul
ret = a @ b
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/umap/distances.py:1063: NumbaDeprecationWarning: The 'nopython' keyword argument was not supplied to the 'numba.jit' decorator. The implicit default value for this argument is currently False, but it will be changed to True in Numba 0.59.0. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@numba.jit()
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/umap/distances.py:1071: NumbaDeprecationWarning: The 'nopython' keyword argument was not supplied to the 'numba.jit' decorator. The implicit default value for this argument is currently False, but it will be changed to True in Numba 0.59.0. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@numba.jit()
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/umap/distances.py:1086: NumbaDeprecationWarning: The 'nopython' keyword argument was not supplied to the 'numba.jit' decorator. The implicit default value for this argument is currently False, but it will be changed to True in Numba 0.59.0. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@numba.jit()
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/umap/umap_.py:660: NumbaDeprecationWarning: The 'nopython' keyword argument was not supplied to the 'numba.jit' decorator. The implicit default value for this argument is currently False, but it will be changed to True in Numba 0.59.0. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@numba.jit()
Note that previous X_pca and X_emb (if exists) will be kept and renamed as X_pca_old and X_emb_old
[11]:
adata_orig
[11]:
AnnData object with n_obs × n_vars = 7438 × 25289
obs: 'time_info', 'state_info', 'NeuMon_fate_bias', 'NeuMon_mask', 'progenitor_Ccr7_DC', 'progenitor_Mast', 'progenitor_Meg', 'progenitor_pDC', 'progenitor_Eos', 'progenitor_Lymphoid', 'progenitor_Erythroid', 'progenitor_Baso', 'progenitor_Neutrophil', 'progenitor_Monocyte', 'sp_500_idx', 'MLPClassifier_predicted_bias', 'n_counts'
var: 'highly_variable'
uns: 'available_map', 'clonal_time_points', 'data_des', 'progenitor_Monocyte_colors', 'state_info_colors', 'time_ordering', 'neighbors', 'umap'
obsm: 'X_clone', 'X_emb', 'X_pca', 'X_pca_old', 'X_umap', 'X_emb_old'
obsp: 'distances', 'connectivities'
[12]:
cs.pl.embedding(adata_orig, color="state_info")
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
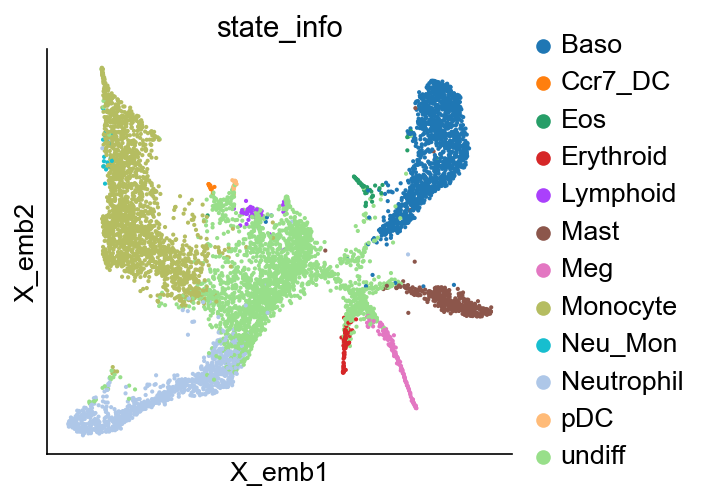
State annotation¶
This generates adata_orig.obs['state_info'].
[13]:
cs.pp.get_state_info(adata_orig, n_neighbors=20, resolution=0.5)
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/extmath.py:152: RuntimeWarning: invalid value encountered in matmul
ret = a @ b
[14]:
cs.pl.embedding(adata_orig, color="state_info")
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
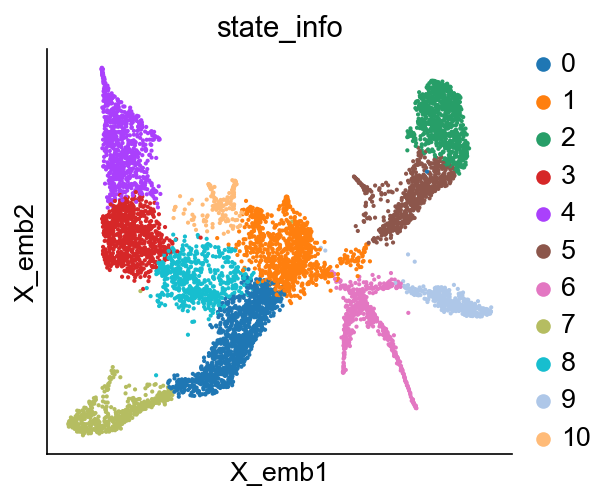
Similarly, the pre-existing state_info will be renamed
[15]:
adata_orig
[15]:
AnnData object with n_obs × n_vars = 7438 × 25289
obs: 'time_info', 'state_info', 'NeuMon_fate_bias', 'NeuMon_mask', 'progenitor_Ccr7_DC', 'progenitor_Mast', 'progenitor_Meg', 'progenitor_pDC', 'progenitor_Eos', 'progenitor_Lymphoid', 'progenitor_Erythroid', 'progenitor_Baso', 'progenitor_Neutrophil', 'progenitor_Monocyte', 'sp_500_idx', 'MLPClassifier_predicted_bias', 'n_counts', 'leiden', 'state_info_old'
var: 'highly_variable'
uns: 'available_map', 'clonal_time_points', 'data_des', 'progenitor_Monocyte_colors', 'state_info_colors', 'time_ordering', 'neighbors', 'umap', 'leiden'
obsm: 'X_clone', 'X_emb', 'X_pca', 'X_pca_old', 'X_umap', 'X_emb_old'
obsp: 'distances', 'connectivities'
Refine state annotation by marker genes¶
The goal here is to refine adata_orig.obs['state_info']. In this method, a state is selected if it expresses all genes in the list of marker_genes, and the expression is above the relative threshold express_threshold. You can also specify which time point you want to focus on. In addition, we also include cell states neighboring to these valid states to smooth the selection (controlled by add_neighbor_N). First, explore parameters to find satisfactory annotation.
[16]:
confirm_change = False
marker_genes = ["Mpo", "Elane", "S100a8"]
cs.pp.refine_state_info_by_marker_genes(
adata_orig,
marker_genes,
express_threshold=0.1,
selected_key="time_info",
selected_values=["4"],
new_cluster_name="new",
add_neighbor_N=10,
confirm_change=confirm_change,
)
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/extmath.py:152: RuntimeWarning: invalid value encountered in matmul
ret = a @ b
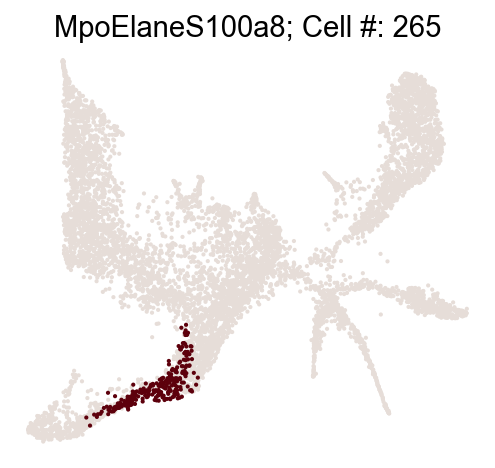
Once you are happy with the result, set confirm_change=True to confirm changes to adata.obs['state_info'].
[17]:
confirm_change = True
marker_genes = ["Mpo", "Elane", "S100a8"]
cs.pp.refine_state_info_by_marker_genes(
adata_orig,
marker_genes,
express_threshold=0.1,
selected_key = "time_info",
selected_values=["4"],
new_cluster_name="new",
add_neighbor_N=10,
confirm_change=True,
)
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/sklearn/utils/extmath.py:152: RuntimeWarning: invalid value encountered in matmul
ret = a @ b
Change state annotation at adata.obs['state_info']
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
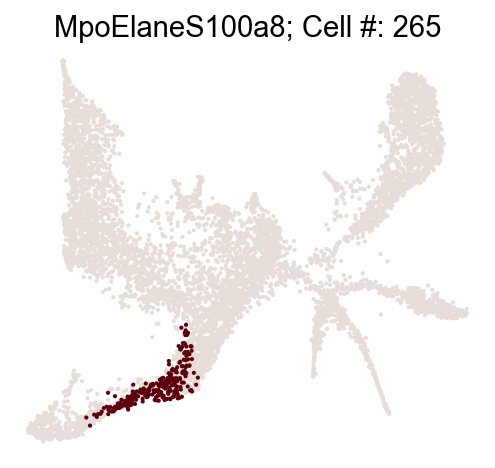
Refine state annotation by clustering states at given time points¶
First, explore the parameters.
[18]:
confirm_change = False
cs.pp.refine_state_info_by_leiden_clustering(
adata_orig,
selected_key="time_info",
selected_values=["2"],
n_neighbors=20,
resolution=0.5,
confirm_change=confirm_change,
)
/Users/shouwen/opt/miniconda3/envs/cospar1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
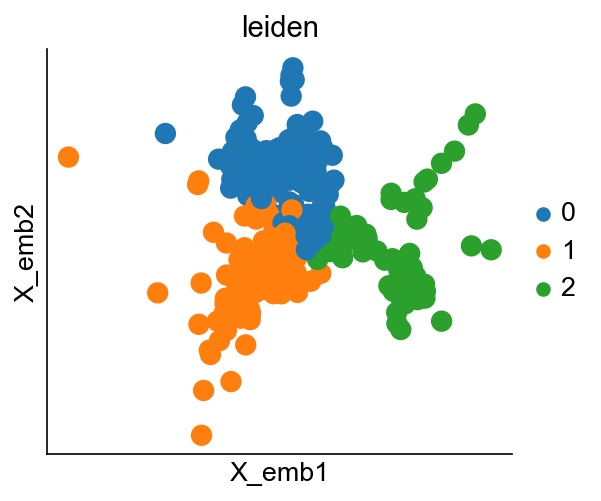
Once you are happy with the result, set confirm_change=True to confirm changes to adata.obs['state_info'].
Miscellaneous¶
[19]:
cs.hf.check_available_choices(adata_orig)
Available transition maps: []
Available clusters: ['9', '3', '0', '7', '2', '10', '4', '6', '5', '1', 'new', '8']
Available time points: ['2' '4' '6']
Clonal time points: ['2' '4' '6']
You can choose to save preprocessed data. It can be loaded using cs.hf.read(file_name).
[20]:
# cs.hf.save_preprocessed_adata(adata_orig)
[ ]: